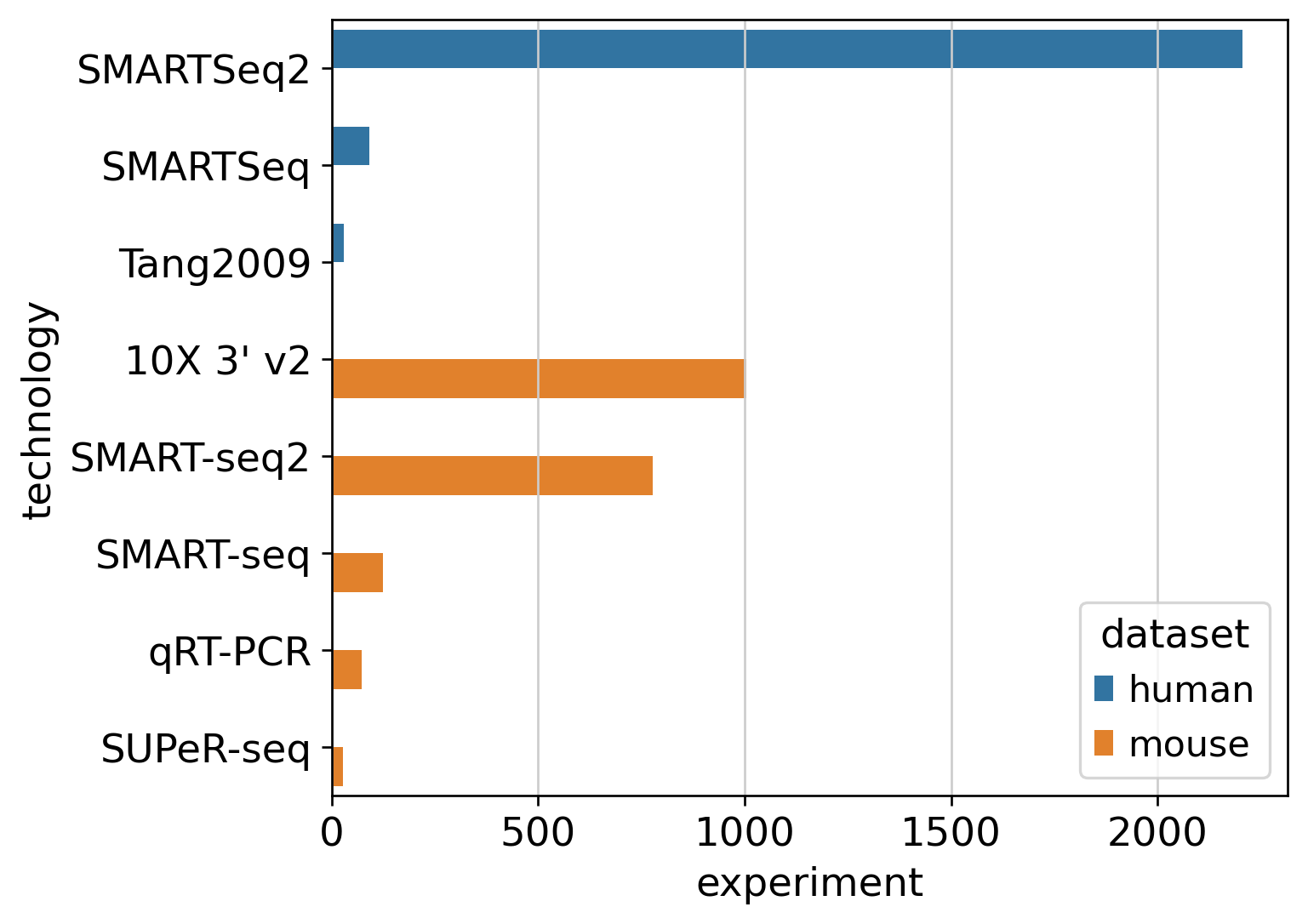
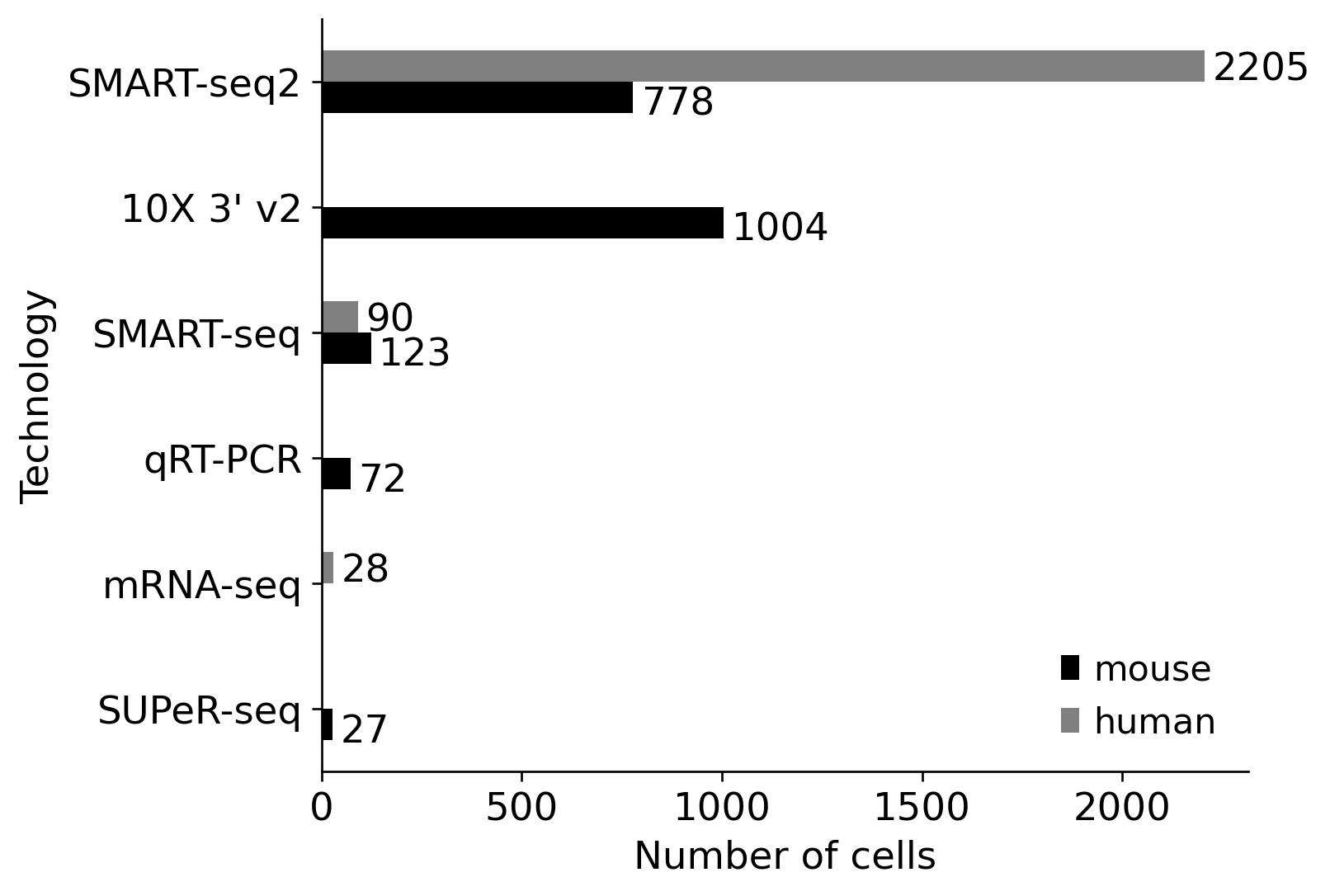
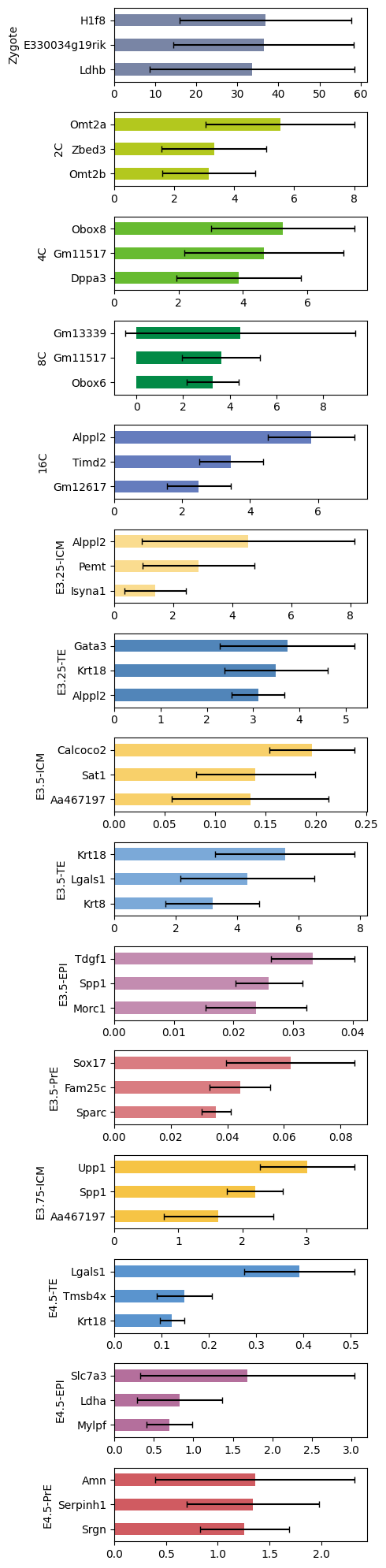
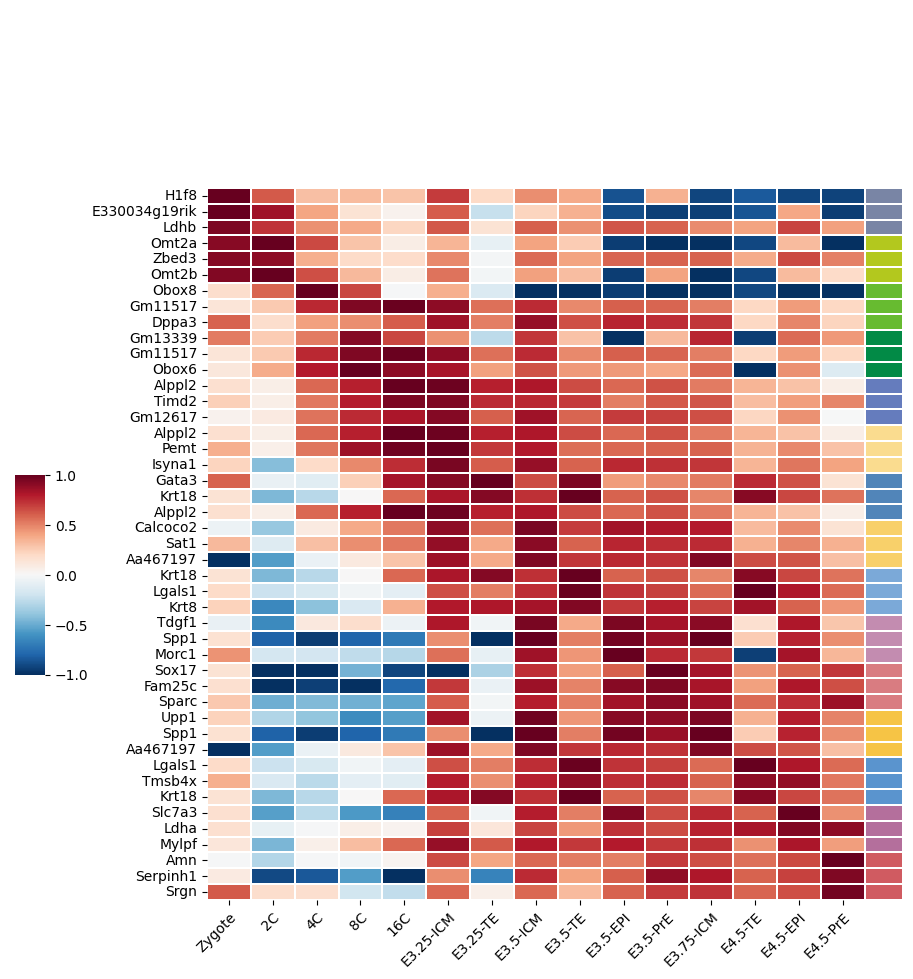
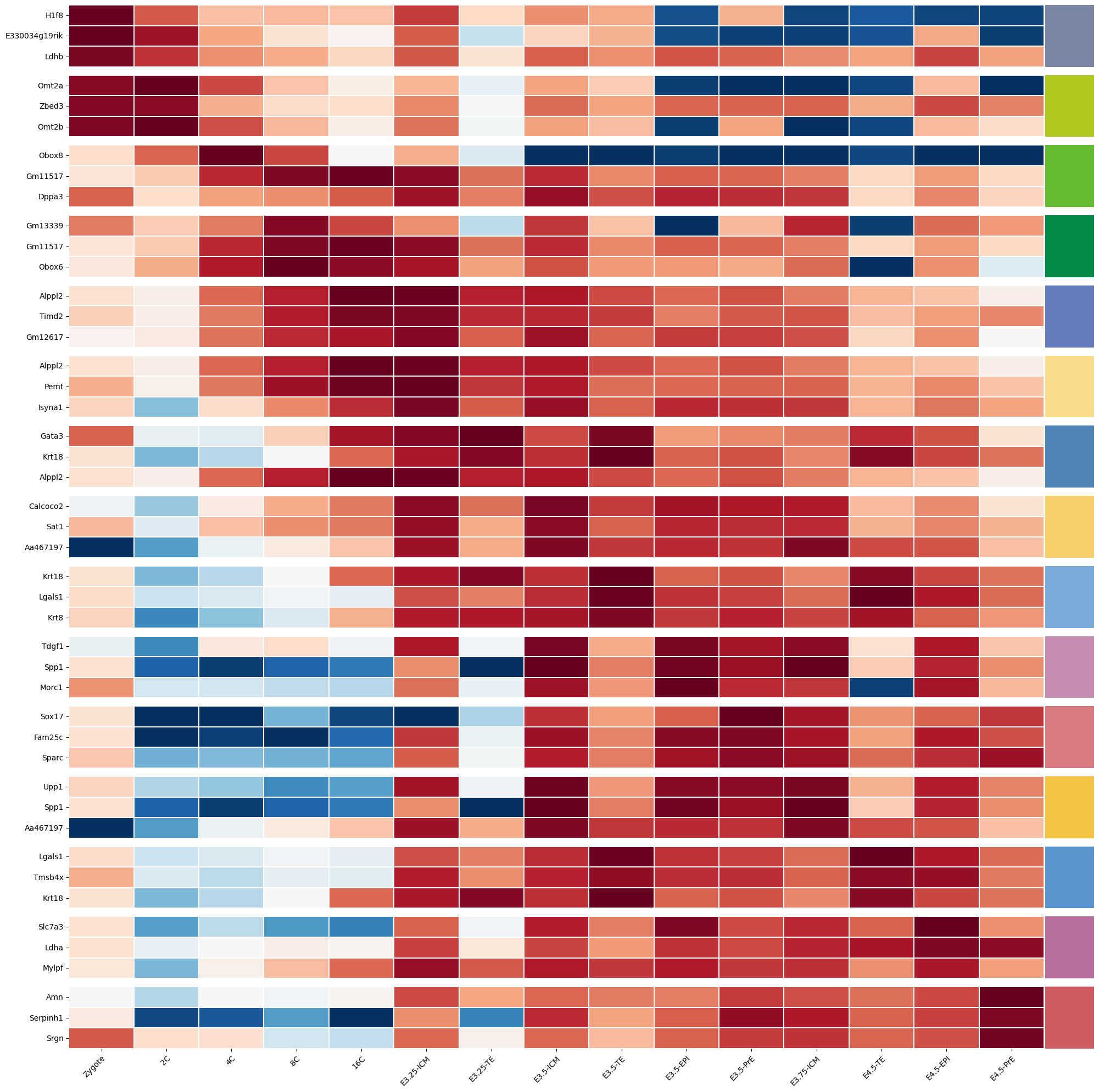
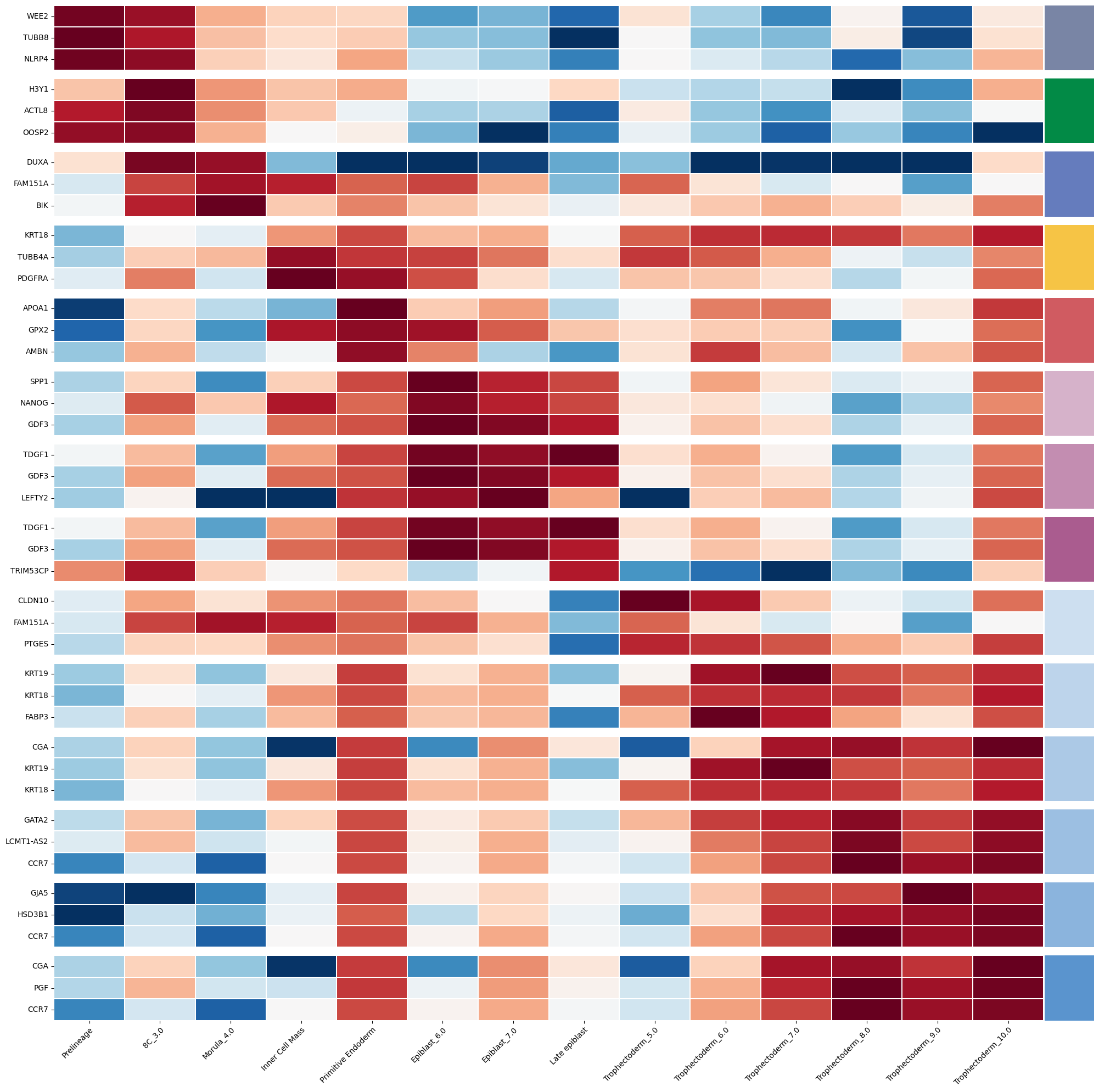
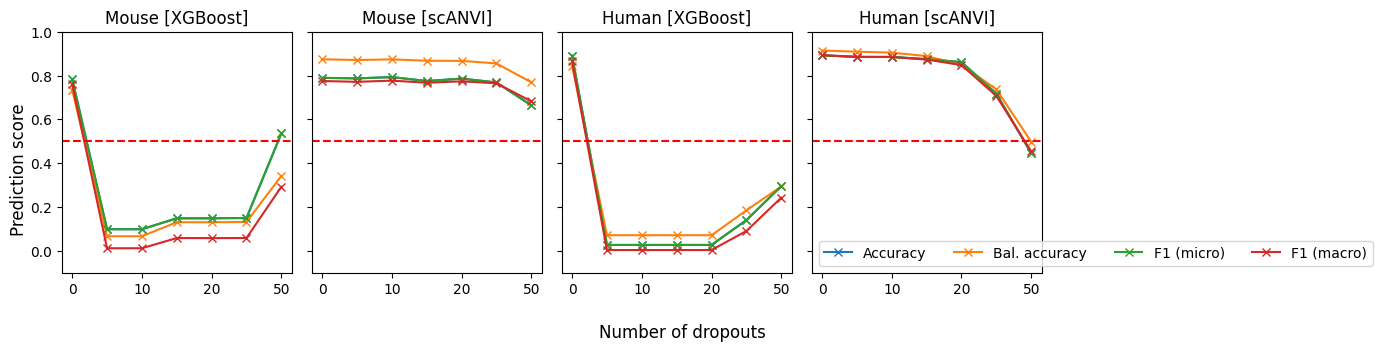
%matplotlib inlineimport numpy as npimport pandas as pdimport seaborn as snsimport scvifrom scvi.model import SCANVIfrom sklearn.preprocessing import MinMaxScalerimport scanpy as scimport cellrank as crimport matplotlib.pyplot as pltimport warningswarnings.simplefilter("ignore", category=UserWarning)
/projects/dan1/data/Brickman/conda/envs/scvi-1.0.0/lib/python3.10/site-packages/scvi/_settings.py:63: UserWarning: Since v1.0.0, scvi-tools no longer uses a random seed by default. Run `scvi.settings.seed = 0` to reproduce results from previous versions.
self.seed = seed
/projects/dan1/data/Brickman/conda/envs/scvi-1.0.0/lib/python3.10/site-packages/scvi/_settings.py:70: UserWarning: Setting `dl_pin_memory_gpu_training` is deprecated in v1.0 and will be removed in v1.1. Please pass in `pin_memory` to the data loaders instead.
self.dl_pin_memory_gpu_training = (
lvae_mouse.adata.obs.predictions = lvae_mouse.adata.obs.predictions.cat.reorder_categories(mouse_ct_colors.keys())lvae_human.adata.obs.predictions = lvae_human.adata.obs.predictions.cat.reorder_categories(list(human_ct_colors.keys())[:-1])lvae_mouse.adata.uns['predictions_colors'] = [mouse_ct_colors[ct] for ct in lvae_mouse.adata.obs.predictions.cat.categories]lvae_human.adata.uns['predictions_colors'] = [human_ct_colors[ct] for ct in lvae_human.adata.obs.predictions.cat.categories]