% matplotlib inlineimport numpy as npimport pandas as pdimport seaborn as snsimport scviimport scanpy as scimport scanpy.external as sceimport scFates as scfimport matplotlib.pyplot as pltimport warningsfrom numba.core.errors import NumbaDeprecationWarning'ignore' , category= NumbaDeprecationWarning)'ignore' , category= FutureWarning )'ignore' , category= UserWarning )
/projects/dan1/data/Brickman/conda/envs/scvi-1.0.0/lib/python3.10/site-packages/scvi/_settings.py:63: UserWarning: Since v1.0.0, scvi-tools no longer uses a random seed by default. Run `scvi.settings.seed = 0` to reproduce results from previous versions.
self.seed = seed
/projects/dan1/data/Brickman/conda/envs/scvi-1.0.0/lib/python3.10/site-packages/scvi/_settings.py:70: UserWarning: Setting `dl_pin_memory_gpu_training` is deprecated in v1.0 and will be removed in v1.1. Please pass in `pin_memory` to the data loaders instead.
self.dl_pin_memory_gpu_training = (
= (10 , 6 ))% config InlineBackend.print_figure_kwargs= {'facecolor' : "w" }% config InlineBackend.figure_format= 'retina'
Rivron dataset SRR filtering
= pd.read_csv('../data/external/kagawa_samplesheet.csv' )
= pd.read_csv('../data/external/kagawa_exclude.txt' , header= None )
= pd.read_csv('../data/external/kagawa_include.txt' , header= None )
= kagawa_samplesheet.loc[kagawa_samplesheet['sample' ].isin(kagawa_include[0 ])].copy()
'../data/external/kagawa_samplesheet_filtered.csv' ,= None ,= csv.QUOTE_NONNUMERIC)
fetch-ngs
~/Brickman/helper-scripts/nf-core_tower.sh Kagawa_2022 nextflow run brickmanlab/scrnaseq \
-r feature/smartseq \
-c /projects/dan1/data/Brickman/projects/proks-salehin-et-al-2023/pipeline/smartseq.human.config \
--input /scratch/Brickman/pipelines/Kagawa_2022/results/samplesheet/samplesheet_filtered.csv
Rivron dataset preprocessing
= sc.read('../data/external/aligned/human/rivron_2022_reprocessed.h5ad' )
AnnData object with n_obs × n_vars = 2673 × 62754
obs: 'sample', 'fastq_1', 'fastq_2', 'run_accession', 'experiment_accession', 'sample_accession', 'secondary_sample_accession', 'study_accession', 'secondary_study_accession', 'submission_accession', 'run_alias', 'experiment_alias', 'sample_alias', 'study_alias', 'library_layout', 'library_selection', 'library_source', 'library_strategy', 'instrument_model', 'instrument_platform', 'scientific_name', 'sample_title', 'experiment_title', 'study_title', 'sample_description', 'fastq_md5', 'fastq_bytes', 'fastq_ftp', 'fastq_galaxy', 'fastq_aspera'
var: 'gene_symbol'
= pd.read_table("../data/external/human/Homo_sapiens.GRCh38.110.gene_length.tsv" , index_col= 0 )= gtf[['median' ]].copy()= ['length' ]def normalize_smartseq(adata: sc.AnnData, gene_len: pd.DataFrame) -> sc.AnnData:print ("SMART-SEQ: Normalization" )= adata.var_names.intersection(gene_len.index)print (f"SMART-SEQ: Common genes { common_genes. shape[0 ]} " )= gene_len.loc[common_genes, "length" ].values= sc.AnnData(adata[:, common_genes].X, obs= adata.obs, dtype= np.float32)= common_genes= normalized.X / lengths * np.median(lengths)= np.rint(normalized.X)return normalized
SMART-SEQ: Normalization
SMART-SEQ: Common genes 62663
AnnData object with n_obs × n_vars = 2673 × 62663
obs: 'sample', 'fastq_1', 'fastq_2', 'run_accession', 'experiment_accession', 'sample_accession', 'secondary_sample_accession', 'study_accession', 'secondary_study_accession', 'submission_accession', 'run_alias', 'experiment_alias', 'sample_alias', 'study_alias', 'library_layout', 'library_selection', 'library_source', 'library_strategy', 'instrument_model', 'instrument_platform', 'scientific_name', 'sample_title', 'experiment_title', 'study_title', 'sample_description', 'fastq_md5', 'fastq_bytes', 'fastq_ftp', 'fastq_galaxy', 'fastq_aspera'
= adata_rivron.obs
= metadata_rivron.loc[:,['sample' ]]
'sample_title' ] = metadata_rivron.sample_title.str .extract(r'^(.*)-' , expand = False )
array(['primed H9', 'blastoid 96h TROP2 pl', 'naive H9', 'okae bts5',
'blastoid 24h', 'blastoid 60h TROP2 pl', 'blastoid 60h TROP2 min',
'blastoid 96h DN', 'blastoid 96h PDGFRa pl',
'blastoid 60h PDGFRa pl'], dtype=object)
= metadata_rivron_clean.loc[metadata_rivron_clean.sample_title.isin(['blastoid 96h TROP2 pl' , 'naive H9' , 'blastoid 24h' , 'blastoid 60h TROP2 pl' , 'blastoid 60h TROP2 min' , 'blastoid 96h DN' , 'blastoid 96h PDGFRa pl' , 'blastoid 60h PDGFRa pl' ])].copy()
'batch' ] = 'Rivron' 'time' ] = metadata_rivron_clean['sample_title' ]'flow' ] = metadata_rivron_clean['sample_title' ]
= {'naive H9' : '0h' ,'blastoid 24h' : '24h' ,'blastoid 60h TROP2 pl' : '60h' ,'blastoid 60h TROP2 min' : '60h' ,'blastoid 60h PDGFRa pl' : '60h' ,'blastoid 96h DN' : '96h' ,'blastoid 96h PDGFRa pl' : '96h' ,'blastoid 96h TROP2 pl' : '96h' = {'naive H9' : 'naive' ,'blastoid 24h' : 'na' ,'blastoid 60h TROP2 pl' : 'TROP2+' ,'blastoid 60h TROP2 min' : 'TROP2-' ,'blastoid 60h PDGFRa pl' : 'PDGFRA+' ,'blastoid 96h DN' : 'Double-neg' ,'blastoid 96h PDGFRa pl' : 'PDGFRA+' ,'blastoid 96h TROP2 pl' : 'TROP2+' = metadata_rivron_clean.replace({'time' : time_replace_dict, 'flow' : flow_replace_dict})
= adata_rivron[metadata_rivron_clean.index].copy()
= metadata_rivron_clean
AnnData object with n_obs × n_vars = 2421 × 62754
obs: 'sample', 'sample_title', 'batch', 'time', 'flow'
var: 'gene_symbol'
'mt' ] = adata_rivron.var.gene_symbol.str .startswith('MT-' )
= ['mt' ], percent_top= None , log1p= False , inplace= True )
= adata_rivron.obs['pct_counts_mt' ], orient= 'v' )
<Axes: ylabel='pct_counts_mt'>
SRX11129000_SRX11129000
SRX11129000
blastoid 96h TROP2 pl
Rivron
96h
TROP2+
2897
1081299.0
450085.0
41.624470
SRX11128994_SRX11128994
SRX11128994
blastoid 96h TROP2 pl
Rivron
96h
TROP2+
6437
2058883.0
172783.0
8.392075
SRX11129132_SRX11129132
SRX11129132
naive H9
Rivron
0h
naive
6808
2237799.0
217539.0
9.721114
SRX11129146_SRX11129146
SRX11129146
naive H9
Rivron
0h
naive
7966
1798534.0
115297.0
6.410610
SRX11129013_SRX11129013
SRX11129013
blastoid 96h TROP2 pl
Rivron
96h
TROP2+
6160
1677046.0
143402.0
8.550869
...
...
...
...
...
...
...
...
...
...
SRX11131981_SRX11131981
SRX11131981
blastoid 60h TROP2 min
Rivron
60h
TROP2-
43
4086.0
0.0
0.000000
SRX11131985_SRX11131985
SRX11131985
blastoid 60h TROP2 min
Rivron
60h
TROP2-
5215
837410.0
196307.0
23.442162
SRX11131982_SRX11131982
SRX11131982
blastoid 60h TROP2 min
Rivron
60h
TROP2-
8915
1567157.0
90066.0
5.747095
SRX11131984_SRX11131984
SRX11131984
blastoid 60h TROP2 min
Rivron
60h
TROP2-
5978
1709532.0
0.0
0.000000
SRX11131980_SRX11131980
SRX11131980
blastoid 60h TROP2 min
Rivron
60h
TROP2-
7449
1845767.0
67667.0
3.666064
2421 rows × 9 columns
= 'total_counts' , y= 'n_genes_by_counts' , data= adata_rivron.obs, hue= 'batch' )
<Axes: xlabel='total_counts', ylabel='n_genes_by_counts'>
= adata_rivron[adata_rivron.obs.pct_counts_mt < 12.5 ].copy()= 2.5e5 )= 2.5e6 )= 2_000 )"counts" ] = adata_rivron.X.copy()= 10_000 )= adata_rivron
# remove mitochondrial genes = adata_rivron[:, adata_rivron.var[~ adata_rivron.var.gene_symbol.str .startswith('MT-' )].index].copy()# remove ribosomal genes = adata_rivron[:, adata_rivron.var[~ adata_rivron.var.gene_symbol.str .startswith(('RPS' , 'RPL' ))].index].copy()
'../results/06_human_Rivron.h5ad' )
= sc.read_h5ad('../results/06_human_Rivron.h5ad' )'experiment' ] = 'Rivron'
AnnData object with n_obs × n_vars = 1762 × 61002
obs: 'sample', 'sample_title', 'batch', 'time', 'flow', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'n_counts', 'n_genes', 'experiment'
var: 'gene_symbol', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
uns: 'log1p'
layers: 'counts'
= scvi.model.SCANVI.load("../results/02_human_integration/05_scanvi_ns15/" )#lvae = scvi.model.SCANVI.load("../results/deprecated/human_integration/version_1/scanvi/")
INFO File ../results/02_human_integration/05_scanvi_ns15/model.pt already downloaded
INFO Found 92.96666666666667% reference vars in query data.
= scvi.model.SCANVI.load_query_data(query, lvae)
= 100 ,= dict (weight_decay= 0.0 ),= 10 ,= True
INFO Training for 100 epochs.
Epoch 100/100: 100%|█████████████████████| 100/100 [00:20<00:00, 4.97it/s, v_num=1, train_loss_step=4.93e+3, train_loss_epoch=4.65e+3]Epoch 100/100: 100%|█████████████████████| 100/100 [00:20<00:00, 4.83it/s, v_num=1, train_loss_step=4.93e+3, train_loss_epoch=4.65e+3]
GPU available: True (cuda), used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
HPU available: False, using: 0 HPUs
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0,1,2,3]
`Trainer.fit` stopped: `max_epochs=100` reached.
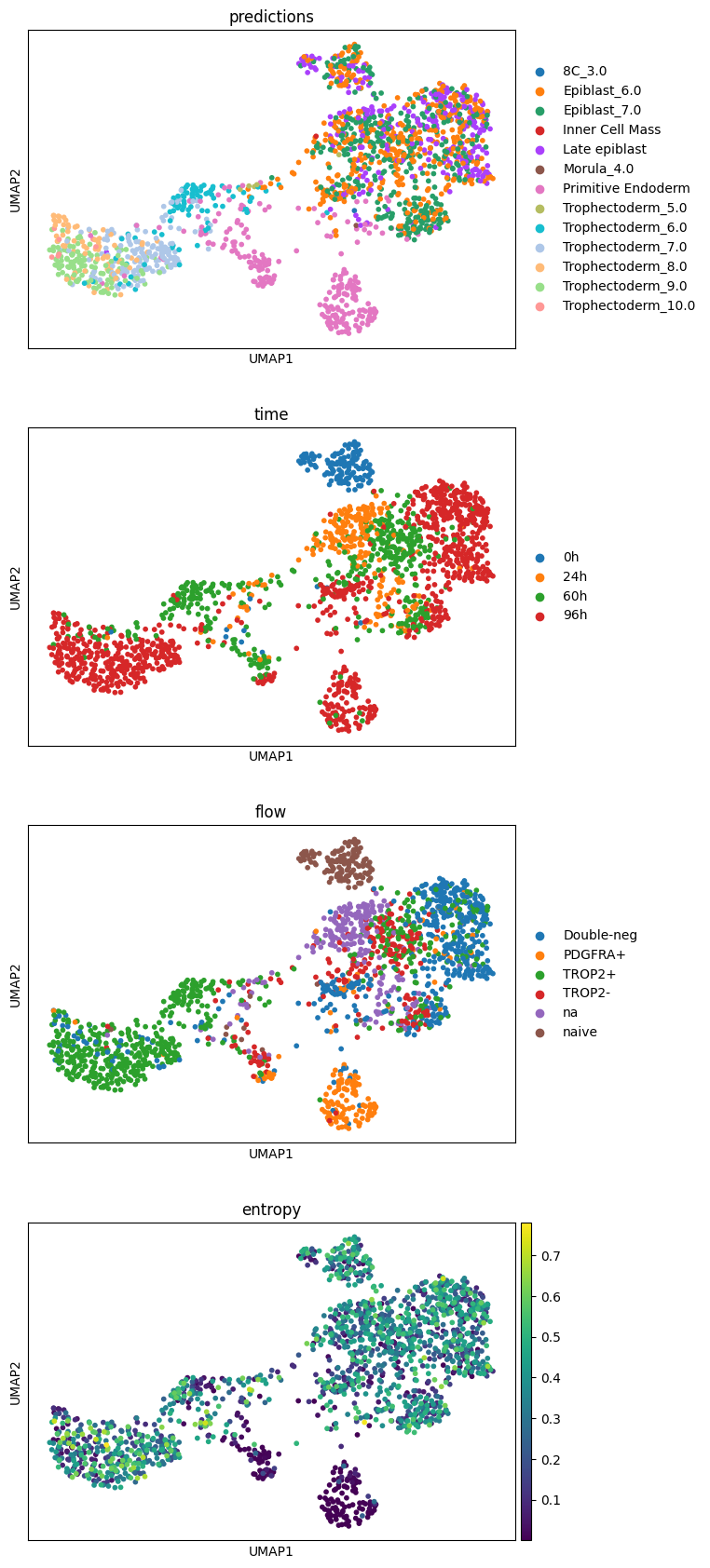
"X_scANVI" ] = lvae_q.get_latent_representation()"predictions" ] = lvae_q.predict()'entropy' ] = 1 - lvae_q.predict(soft= True ).max (axis= 1 )
predictions
8C_3.0
1
0
0
0
0
0
Epiblast_6.0
146
8
52
80
72
49
Epiblast_7.0
120
9
71
65
88
39
Inner Cell Mass
0
1
0
0
0
0
Late epiblast
124
12
42
18
32
42
Morula_4.0
1
0
0
0
0
0
Primitive Endoderm
33
121
33
36
16
6
Trophectoderm_10.0
3
0
4
0
0
0
Trophectoderm_5.0
0
0
1
0
4
0
Trophectoderm_6.0
4
0
76
2
6
1
Trophectoderm_7.0
15
0
140
1
1
0
Trophectoderm_8.0
14
2
52
0
0
1
Trophectoderm_9.0
7
0
111
0
0
0
predictions
8C_3.0
0
0
0
1
Epiblast_6.0
49
72
120
166
Epiblast_7.0
39
88
111
154
Inner Cell Mass
0
0
1
0
Late epiblast
42
32
38
158
Morula_4.0
0
0
0
1
Primitive Endoderm
6
16
66
157
Trophectoderm_10.0
0
0
0
7
Trophectoderm_5.0
0
4
1
0
Trophectoderm_6.0
1
6
62
20
Trophectoderm_7.0
0
1
30
126
Trophectoderm_8.0
1
0
4
64
Trophectoderm_9.0
0
0
5
113
= "seurat_v3" ,= 5_000 ,= "counts" ,= "batch" ,= True ,
WARNING: You’re trying to run this on 3000 dimensions of `.X`, if you really want this, set `use_rep='X'`.
Falling back to preprocessing with `sc.pp.pca` and default params.
2024-01-12 17:05:15.412966: W tensorflow/compiler/tf2tensorrt/utils/py_utils.cc:38] TF-TRT Warning: Could not find TensorRT
= ['predictions' ,'time' , 'flow' , 'entropy' ], ncols= 1 )