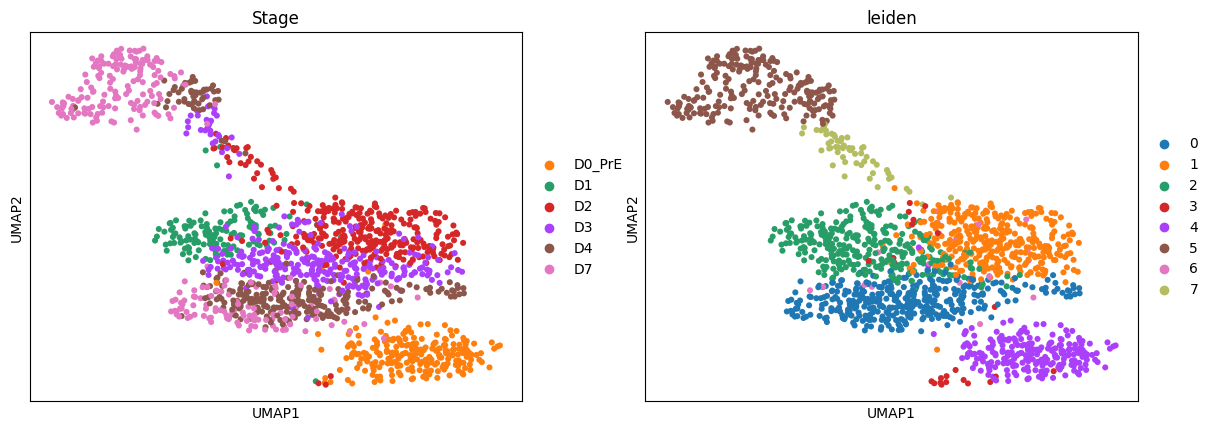
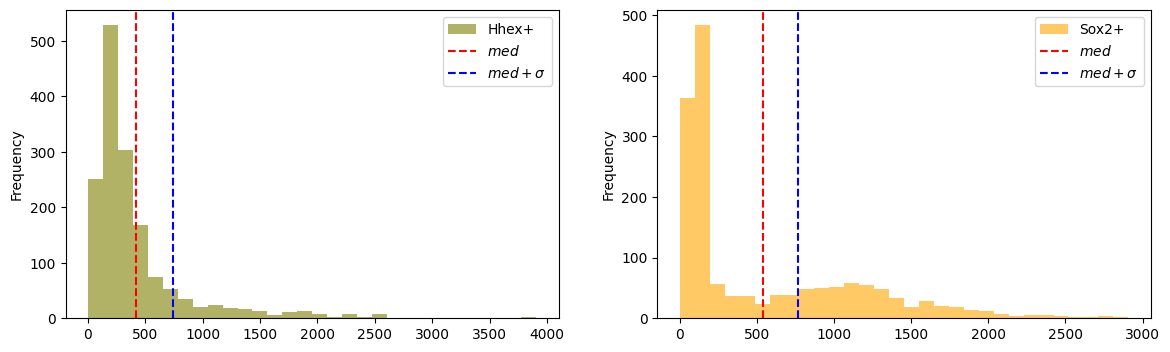
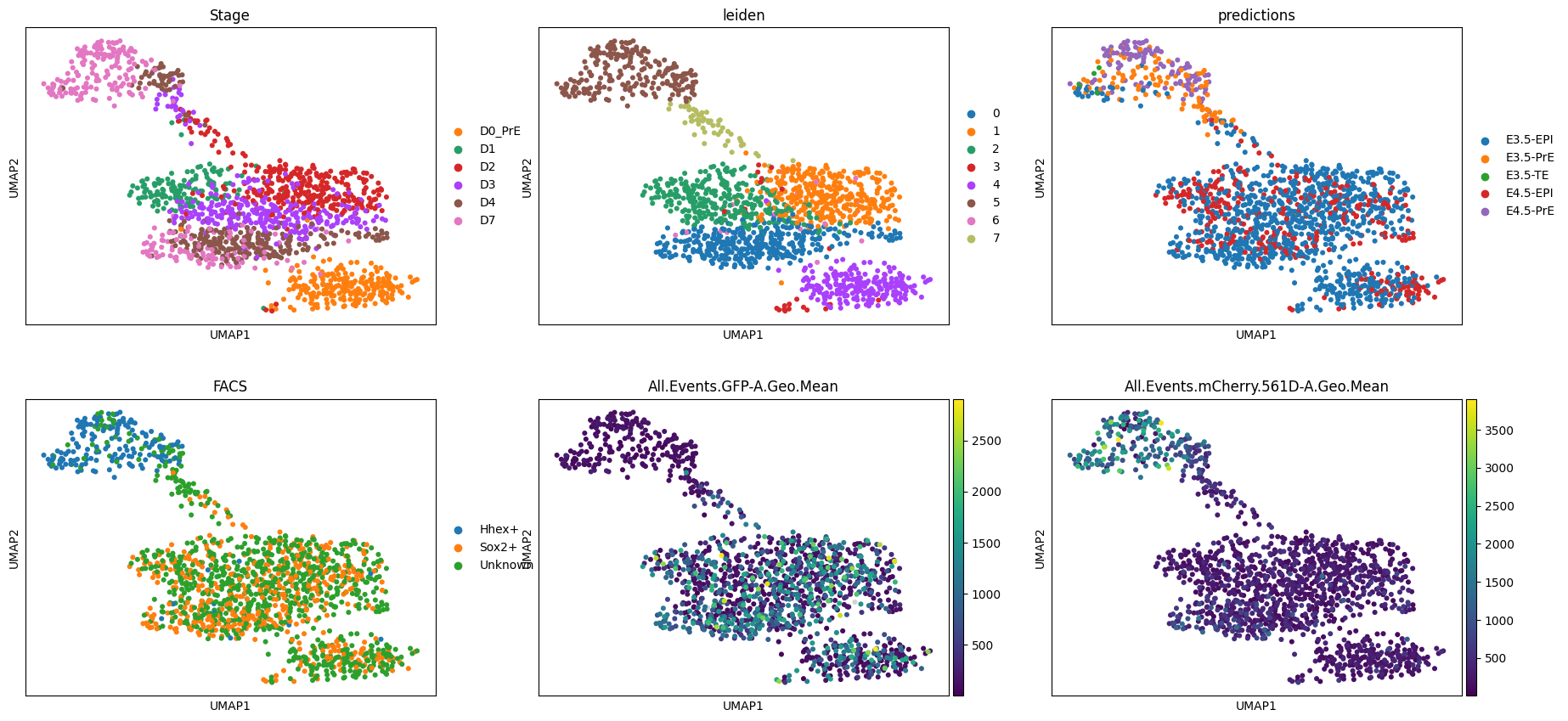
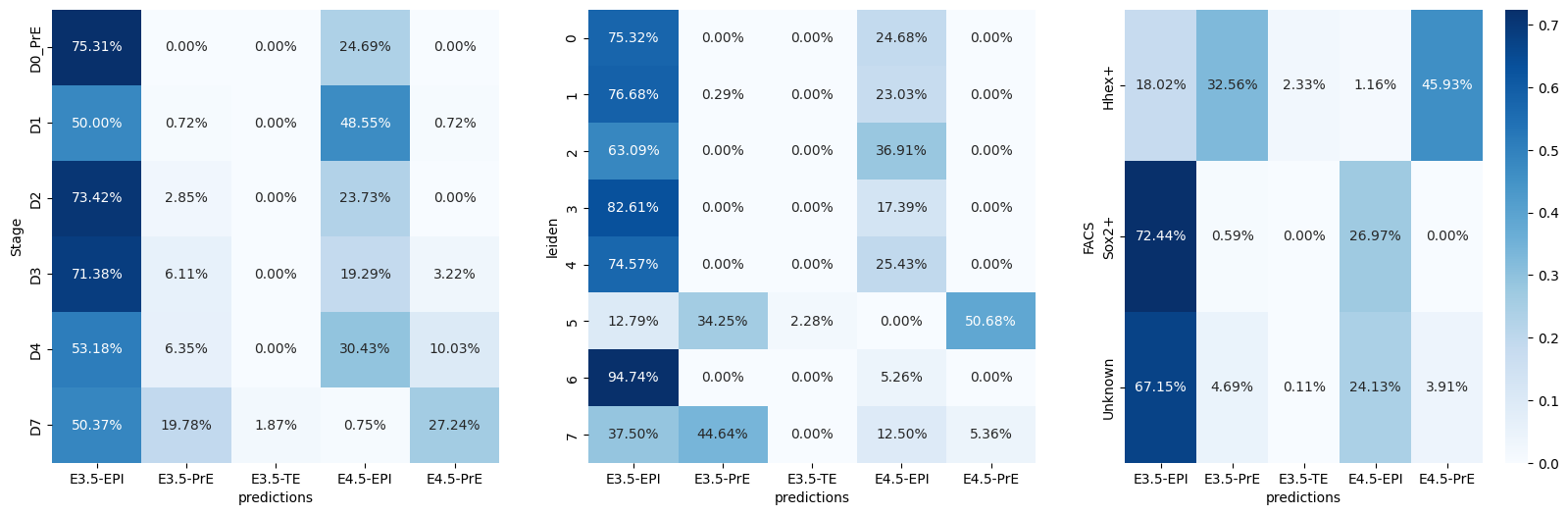
AnnData object with n_obs × n_vars = 1575 × 20459
obs: 'Well_coordinates', 'plate_ID', 'Amp_batch_ID', 'Cell_barcode', 'Pool_barcode', 'Batch', 'Condition', 'SampleName', 'Stage', 'Source', 'Day', 'Index sort', 'All.Events.FSC-A.Geo.Mean', 'All.Events.SSC-A.Geo.Mean', 'All.Events.GFP-A.Geo.Mean', 'All.Events.mCherry.561D-A.Geo.Mean', 'All.Events.DAPI-A.Geo.Mean', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'total_counts_ercc', 'pct_counts_ercc', 'total_counts_ribo', 'pct_counts_ribo', 'n_genes', 'n_counts', 'S_score', 'G2M_score', 'phase', 'louvain', 'leiden', 'umap_density_Stage', 'batch', 'initial_size_spliced', 'initial_size_unspliced', 'initial_size', 'velocity_self_transition', 'root_cells', 'end_points', 'velocity_pseudotime', 'latent_time', 'velocity_length', 'velocity_confidence', 'velocity_confidence_transition'
var: 'mt', 'ercc', 'ribo', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std', 'gene_ids'
layers: 'counts'